Sébastien Calvignac-Spencer searches museum jars for genetic traces of flu, measles and other viruses. Their evolutionary stories can help treat modern outbreaks and prepare for future ones.
Introduction
Like many children of his generation, raised on a steady diet of Jurassic Park and its sequels, Sébastien Calvignac-Spencer wanted to be a paleontologist when he grew up. Unlike most, he came close. Rather than digging for fossilized bones in the dirt, however, he paws through objects in natural history museums and medical collections for old biological specimens from which he can extract the genomes of disease-causing viruses. He hopes such old RNA molecules can bring us closer to understanding how pathogens evolve. “Instead of just inferring things and making educated guesses, we could actually observe evolution,” he said.
His first efforts toward this goal didn’t go exactly as planned. In his early 20s, Calvignac-Spencer went looking for simian immunodeficiency viruses in century-old monkey bones from a natural history museum. But to his disappointment, he failed to find them. So he decided to focus on modern samples for a while. He studied the origins of the Ebola virus that caused an outbreak in West Africa in 2014. Then he investigated malaria and leprosy in wild apes. And when SARS-CoV-2 kicked off a global pandemic, he helped set up genomic surveillance of the virus at the Robert Koch Institute in Berlin.
These contemporary experiences only deepened his resolve to study the genomes of historical viruses. Calvignac-Spencer and his colleagues often wanted to know how the pathogens they studied had emerged and evolved. To find out, they’d need to compare modern genomes with ancient genetic samples to reconstruct evolutionary history. “There’s only so much that we can reconstruct from the information we can obtain today,” he said. A historical sample would have been a gift.
By the late 2010s, with the arrival of new technologies such as high-throughput genome sequencing, which is faster and less expensive than prior methods, studying old viral genomes on a larger scale finally appeared feasible. Before long, Calvignac-Spencer obtained the oldest known measles genome from a lung preserved in 1912. He discovered that the virus’s genome has remained surprisingly stable over time — a fact that could help researchers eradicate measles altogether. In 2022, he published a complete genome of Spanish flu from a lung taken from a 17-year-old girl who died of pneumonia in 1918. From this genome, he learned that our seasonal H1N1 influenza virus is likely a direct descendant of that pandemic-causing pathogen.
Today Calvignac-Spencer leads the department of pathogen evolution at the Helmholtz Institute for One Health in Germany, where he analyzes bones from natural history museums and formalin-fixed specimens from medical collections to locate accidentally preserved viral RNA. Quanta spoke with him about the challenges of extracting genetic material from century-old specimens and how bygone viral RNA can help us understand the arms race between humans and pathogens. The interview has been condensed and edited for clarity.
You’ve worked quite a lot on viral outbreaks in African wild animals, apes in particular. How did this experience change your perspective on studying pathogen genomes?
For a long time, it was believed that to do pathogen genomics, you had to have good-quality samples, such as an isolate — a pure sample of a pathogen — or, in the worst case, a piece of tissue that would be heavily infected by the pathogen. But when you work on wild animals, as I and my colleagues do, you don’t have the luxury of having the perfect sample very often.
The only thing that we may get from living chimps, for example, are feces. Imagine there is a respiratory outbreak. The first thing that comes to mind is not “OK, I will go and fetch a bit of poo and try to sequence the respiratory agent.” But we do that. And we realized there are a lot more places where we can look for a pathogen’s genomic material. It’s much more pervasive and resistant than we used to think.
One such place, as you’ve found, is museum collections. How do you go about locating a particular specimen that may contain valuable viral genomes?
It’s still like looking for a needle in a haystack, to be honest. The pathological collections in which we search for specimens were a big thing in the 19th and early 20th centuries, since they were used to train doctors at medical universities. But they’ve lost their appeal with the rise of alternative didactic tools like photographs. In the ’70s and ’80s, many people felt pathological collections had no value for biological research, and these collections largely fell into disarray. Many specimens were lost, which is a bit disheartening. Today, many collections don’t have strong institutional support, so updated catalogs are rare, and in some cases there is no catalog at all.
You’ve still managed to find several needles in that haystack — for example, the formalin-fixed lung sample of a 17-year-old girl who died in Munich in 1918, which contained a complete genome of Spanish influenza. How did you do that?
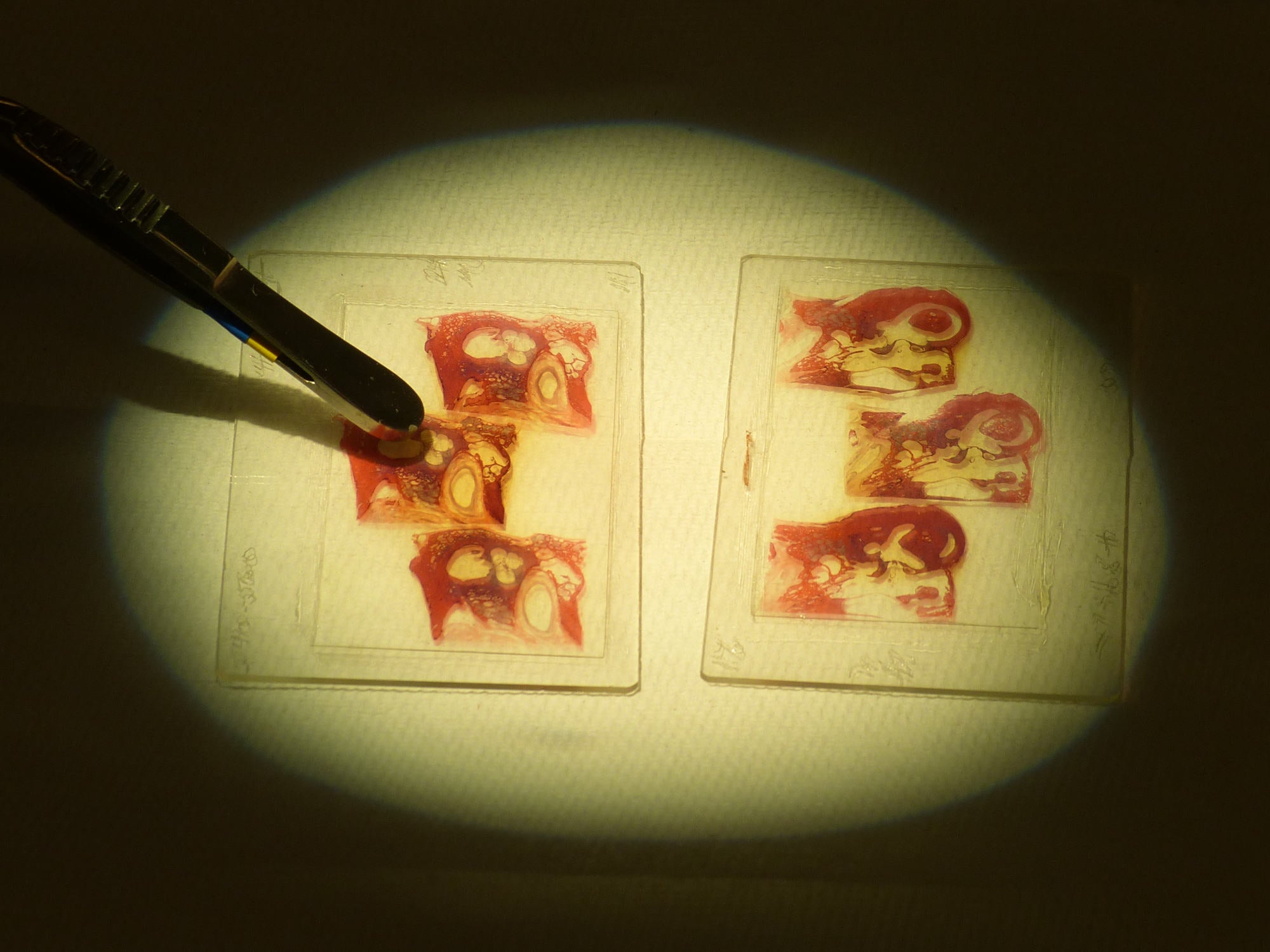
We found that specimen in the pathology collection of the Berlin Museum of Medical History at the Charité. When we first visited, they did not know whether they had 1918 flu cases or not. The collection was in the basement, as it usually is. They had approximately 350 lung and respiratory tract specimens. We just looked at every jar, one by one, noting things that we found interesting.
Many of the specimens you study are human organs fixed in formalin that have been kept in glass jars for over 100 years. Once you find a promising specimen, how do you handle it?
We use complete personal protective equipment: overall suits, several pairs of gloves, hairnets, face masks. But it’s not to protect ourselves. It’s to protect the specimen.
You aren’t worried about catching a virus from your samples?
Not at all. Everything inside is dead. Every biological process has been disrupted by formalin. This is why the preparation is so amenable to preserving viral RNA: You put a complete stop to every enzymatic process, including the degradation of RNA. But humans carry respiratory pathogens all the time. When we go to study the 1918 flu, we don’t want to contaminate our samples with contemporary flu.
So, you are wearing protective equipment. The jar is open. What’s next?
In most collections, the curators insist that we don’t remove the specimens from the jars. So we typically work with tools, like scissors, which have specific shapes so that we can cut a small piece of the tissue while leaving the specimen in the jar. We put these samples into small tubes and bring them back to the lab. Then we boil them. It’s a bit counterintuitive, but it helps release more nucleic acid fragments. Exposure to formalin induces cross-links between DNA and RNA and other big molecules, so they become sort of locked to other molecules. But these links are reversible through heat. After boiling the sample for 15 minutes, we mash it and separate the nucleic acid from all other molecules. Then we take the nucleic acids and prepare them for sequencing.
Before you found the 1918 Spanish flu genome, the prevailing hypothesis was that our seasonal H1N1 flu virus formed when genomic segments from different influenza viruses were exchanged between strains. You found something different.
These viruses have segmented genomes, and sometimes they swap parts of their genome. When there is a co-infection [of multiple viruses] in a host, sometimes different segments will be packaged together. It was previously thought that probably one of the segments had been swapped between the 1918 pandemic virus and the seasonal flu. And what our genomic sequences suggest is that, actually, no, it did not happen. We showed that there was this accelerated rate of evolution for the pandemic lineage that led to seasonal flu.

The valuable information Calvignac-Spencer gleaned from the genomes of old flu and measles viruses has validated his approach. “There’s only so much that we can reconstruct from the information we can obtain today,” he said.
Martin Pauer for Quanta Magazine
We have no explanation for why it happened. But when you take that acceleration into account, the evolutionary history we infer becomes compatible with the idea that the eight segments of the pandemic virus would be the progenitors of the eight segments of the seasonal flu. It’s the same lineage.
Did the virus’s evolution accelerate because of the high number of people infected during the pandemic?
It doesn’t mean you will see such an acceleration — something else must be going on. For example, we think that some SARS-CoV-2 variants that have also clearly experienced accelerated rates of evolution may have evolved in specific reservoirs within the human population, like immunosuppressed people. And the more such cases there are, the more opportunities there are for unlikely mutations. It seems that very rare events can have a disproportionate weight in pandemics.
How can your findings about the accelerated rate of viral evolution during the Spanish flu pandemic help us better prepare for future outbreaks?
Evolution never repeats itself, but sometimes it takes similar paths to similar effects. These findings give us a better sense of what the possibilities are. That accelerated evolution of 1918 flu is reminiscent of what we saw with SARS-CoV-2. And maybe that’s something that we should always keep in mind when we study pandemic events.
On the other hand, the measles virus appears to have changed more slowly than previously believed. When you added the 1912 measles genome into an evolutionary model, you discovered that this virus may have emerged more than 1,000 years earlier than anyone thought. Why was this old RNA such a game changer?
We wanted to estimate the age of divergence from its closest relative, rinderpest, a virus that infected cattle and was eradicated through vaccination in 2011. But the 40 to 50 years of genome sampling that we’ve previously had for measles was a very, very shallow period. It’s not reasonable to extrapolate from such a short period to a period that is much, much longer. If you do that, you will systematically underestimate the age of ancient events.
Why is that?
Because viral genomes are really small, changes [genetic mutations] can happen multiple times at the same position. It’s like a self-erasing process, which makes statistical modeling more difficult. And then there’s the fact that substitution rates [frequency of genetic mutations] themselves might not be homogeneous through time: They can accelerate and slow down. That’s why one solution is to sample deeper through time. Now, instead of the oldest measles genome being from the ’60s or ’70s, we have one from the 1910s. When we’ve inputted this ancient genome into a statistical model, we’ve ended up showing that the divergence with rinderpest happened about 2,500 years ago.
Was there anything significant about this specific date?
It coincided almost exactly with the time when very large cities started to pop up in different places around the world. Measles cannot persist unless a critical population size of about a quarter to a half-million people has been reached. It constantly needs new susceptible individuals. It’s a virus that’s very immunogenic — if you get it once, your immune system can block any further infection. That’s why vaccination works so well.
You recently pitted the 1912 virus against antibodies induced by the current measles vaccine. What happened?
We synthesized the measles surface protein based on this ancient genome and checked whether it would be recognized by the antibodies of recently vaccinated people. And it was. It extended the shelf life of the measles vaccine to 100 years, which means it’s probably not a priority to develop new measles vaccines.
So the measles virus evolves very slowly?
Actually, the virus changes quite fast, but it’s evolving in a sort of evolutionary cage: It cannot go beyond it without losing fitness. I’m not saying there will never be a vaccine escape. I’m saying it seems like a very unlikely event. But the more we let measles circulate in human populations, the more chances it has. Eradication is the only safe way to go. Now our study shows we have a tool — the vaccine — that is almost perfect. And if you have the perfect tool to eradicate such a dangerous disease, why would you not use it?
Note: This article have been indexed to our site. We do not claim legitimacy, ownership or copyright of any of the content above. To see the article at original source Click Here

Stanley Quencher H2.0 FlowState Stainless Steel Vacuum Insulated Tumbler with Lid and Straw for Water, Iced Tea or Coffee, Smoothie and More, Rose Quartz 2.0, 30 OZ / 0.89 L
$35.00 (as of December 21, 2024 19:33 GMT +00:00 - More infoProduct prices and availability are accurate as of the date/time indicated and are subject to change. Any price and availability information displayed on [relevant Amazon Site(s), as applicable] at the time of purchase will apply to the purchase of this product.)
Sonic X Shadow Generations - Nintendo Switch
$39.97 (as of December 21, 2024 19:12 GMT +00:00 - More infoProduct prices and availability are accurate as of the date/time indicated and are subject to change. Any price and availability information displayed on [relevant Amazon Site(s), as applicable] at the time of purchase will apply to the purchase of this product.)
Dickies Men's Dri-Tech Moisture Control Crew Socks, Available in M-XXL (6, 12, 18 Pairs)
$10.98 (as of December 21, 2024 19:33 GMT +00:00 - More infoProduct prices and availability are accurate as of the date/time indicated and are subject to change. Any price and availability information displayed on [relevant Amazon Site(s), as applicable] at the time of purchase will apply to the purchase of this product.)
bella 2 Slice Slim Toaster, Fits-anywhere™ Kitchenware, 6 Setting Shade Control with Reheat & Cancel Buttons, Removable Crumb Tray, 10” Long Slot, Anti Jam & Auto Shutoff, 900 Watt, Oatmilk
$19.99 (as of December 21, 2024 19:05 GMT +00:00 - More infoProduct prices and availability are accurate as of the date/time indicated and are subject to change. Any price and availability information displayed on [relevant Amazon Site(s), as applicable] at the time of purchase will apply to the purchase of this product.)